发病机制
发病机制
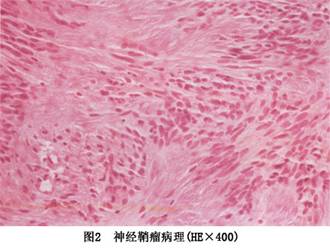
发病机制:肿瘤具有完整的包膜,切面可呈淡红、灰白或黄色。有时可见由变性而形成的囊肿,内含血性液体。镜下见瘤实质主要由神经鞘细胞构成,偶见成熟神经节细胞和神经干参与。根据组织结构特点可分为致密型和网状型两种。
1.致密型(Antoni甲型) 有下列特点:施万细胞通常呈窦状或束条状排列,有细的结缔组织纤维;胞核有呈栅栏状排列倾向,并与无核区相间。
2.网状型(Antoni乙型) 施万细胞排列疏散紊乱,间质水肿。可见基质黏液变性形成多个小囊肿,小囊肿可相互融合形成大囊腔,腔内充满液体。瘤体内可见较多的肥大的细胞。肿瘤内血管丰富,尤其是疏松网状区,血管壁薄,伴有
血栓形成及出血。
临床表现
临床表现:好发于四肢,尤其是下肢。亦可见于头、颈、面、胃、腹腔后及后纵隔等。颅内、椎管内也不太少见。
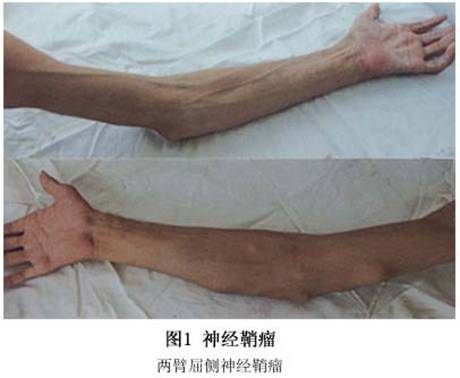
1.坚实结节 为真皮或皮下组织内单个或多个坚实结节,呈圆形或卵圆形,直径通常不超过2~4cm,可推动(图1)。性质柔软或坚实,颜色粉红或淡黄色,可有或无疼痛。偶尔损害呈多发性,可见于Ⅰ型神经纤维瘤病或独立发生,与Ⅰ型神经纤维瘤病无关。但多发性损害常与Ⅱ型神经纤维瘤病伴发。独立型可为先天性或为迟发性。可散发或呈家族性。临床上表现为3种外观,即高起圆屋顶形结节,淡褐色硬结性斑疹和多发性丘疹融合成2~100mm的斑块。
丛状神经鞘瘤(plexiform schwannomas)可为单发性或多发性损害,局限性或泛发性,发生于真皮或皮下组织。可单独发生或与Ⅰ型、Ⅱ型神经纤维瘤病或多发性神经鞘瘤伴发。神经鞘瘤的另一亚型为沙样瘤型与Carney综合征的斑状色素沉着,黏液瘤和内分泌活性过度伴发,可发生恶性亚型,为与Ⅰ型神经纤维瘤病伴发的单发性损害。或在某些病例中与色素性
干皮病伴发。
2.酸麻感 一般无自觉症状,但触之有酸麻感。体积增大时常伴有放射样疼。
3.感觉及运动障碍 如肿瘤显著压迫神经时,可出现感觉及运动障碍。
鉴别诊断
鉴别诊断:
1.神经纤维瘤 神经纤维瘤的组织病理有如下特点:
(1)质韧,有弹性,无包膜。
(2)瘤细胞疏散,栅栏状或旋涡状结构偶见。
(3)瘤体内可见起源的神经常穿插于其中。
(4)瘤体内常可见汗腺及脂肪组织,无厚壁血管。
(5)阿新蓝(Alcian blue)染色阳性。
2.恶性神经鞘瘤与富于细胞的神经鞘瘤要注意鉴别。前者包膜不完整,瘤组织常伴有坏死,常有向周围组织浸润现象;细胞核呈多形性,有明显异形,可见瘤巨细胞,核分裂象易见。
3.硬化型神经鞘瘤似平滑肌瘤或纤维瘤,但多处切片总可找到神经鞘瘤的典型结构。细胞特点及V.G.氏染色有助于鉴别。
4.疼痛时应与血管球瘤鉴别。